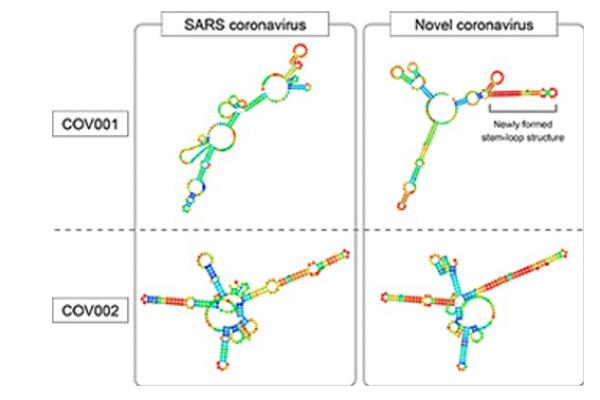
Researchers at the University of Tokyo and University of Tsukuba have identified specific areas of the viruses' genomes that are shared with many other evolutionarily similar viruses. The hairpin shape of these parts of the viruses' genomes may help the viruses avoid a host's immune system. Due to a few important mutations, the genome area named COV001 does not form a hairpin structure in the SARS virus (top left), but does in the COVID-19 virus (top right). First published in Wakida et al. 2020, DOI: 10.1016/j.bbrc.2020.05.008
Researchers have identified specific portions of the genetic codes of the COVID-19 and SARS viruses that may promote the viruses' lifecycles. The new technique is researchers' first tool for determining what genetic sequences stored as RNA -- DNA's chemical cousin -- are more stable.
"The current pandemic is very severe, and we want to contribute to the global acceleration of coronavirus research. Our research includes many types of viruses, but we decided to focus on our coronavirus results," said professor Nobuyoshi Akimitsu, leader of the research team that performed the work and an expert in how cells withstand stress at the University of Tokyo Isotope Science Center.
Many families of viruses - including influenza viruses and coronaviruses - store their genetic sequence as RNA, which sneaks into human cells and tricks them into making more viruses. Viruses need their RNA to remain stable, resisting efforts from the host's immune system to degrade their RNA.
The research team named their technique Fate-seq because it aims to determine the fate of a genetic sequence, whether it will persist or degrade based on its stability.
"The Fate-seq technique is a very simple idea. We combined existing technologies in a new way," explained Akimitsu.
To perform Fate-seq, researchers first cut a genome into short fragments. Even extremely dangerous pathogens become harmless when researchers only work with short, separated, cut-up fragments of their genomes.
Researchers synthesized RNAs from the fragments of virus genomes and examined their fate, that is stability, by using next-generation sequencing, which allows researchers to quickly and simultaneously identify the exact sequence of individual strands of RNA. Computer programs can then identify patterns or interesting differences in the genetic sequences to study in further detail.
Researchers studied 11,848 RNA sequences from 26 virus genomes, including that of SARS-CoV, the virus that causes SARS, the sudden acute respiratory syndrome that killed 774 people in the first half of 2003. Researchers identified a total of 625 stable RNA fragments. Among the stable RNA fragments, 21 were from SARS-CoV.
Researchers compared the 21 SARS-CoV stable genome fragments to the full genetic sequence data available for other types of coronaviruses. Two of the stable fragments from the SARS-CoV are very common in other evolutionarily similar coronaviruses, including the virus that causes COVID-19, SARS-CoV-2.
Predictive models showed that those two stable RNA fragments likely form stem-and-loop structures. Stem-and-loops are short pieces of RNA that, instead of remaining a straight line, fold forward and bind onto themselves, forming a hairpin shape.
Most notably, one of the stable fragments only forms a stem-and-loop in the COVID-19 virus, not the SARS virus due to the few small but important differences in the viruses' RNA codes.
"The stem-and-loop structure of this SARS-CoV-2 genetic fragment is very stable in computer models and we propose that this structure might enhance survival of the virus," said Akimitsu.
In addition to better understanding dangerous viruses, researchers hope to use Fate-seq to understand the fundamental rules of RNA stability and advance new types of medicine. Human cells use RNA as the intermediate messenger between DNA and protein. Designing RNA-based drugs that are stable and easy for cells to translate into protein could treat genetic diseases without the hazards of altering our DNA.
This research was performed with collaborators at the University of Tsukuba and is a peer-reviewed publication in the journal Biochemical and Biophysical Research Communications.
Source: University of Tokyo
Be the first to comment on "Genetic Study Reveals Similarities and Differences of COVID-19 and SARS Viruses"