Researchers from Drexel University are using a new technique for spotting patterns in the genetic sequence of the SARS-CoV-2 coronavirus that can help scientists trace the path of its transmission and how it has mutated.
Drexel University researchers have reported a method to quickly identify and label mutated versions of the virus that causes COVID-19. Their analysis, using information from a global database of genetic information gleaned from coronavirus testing, suggests that there are at least 8 to 14 slightly different versions of the virus infecting people in America, some of which are either the same as, or have subsequently evolved from, strains directly from Asia, while others are the same as those found in Europe.
First developed as a way of parsing genetic samples to get a snapshot of the mix of bacteria, the genetic analysis tool teases out patterns from volumes of genetic information and can identify whether a virus has genetically changed. They can then use the pattern to categorize viruses with small genetic differences using tags called Informative Subtype Markers (ISM).
Applying the same method to process viral genetic data can quickly detect and categorize slight genetic variations in the SARS-CoV-2, the novel coronavirus that causes COVID-19, the group reported in a paper recently published in the journal, PLoS Computational Biology. The genetic analysis tool, designed by Drexel graduate researcher Zhengqiao Zhao, that generates these labels is publicly available for COVID-19 researchers on GitHub.
"The types of SARS-CoV-2 viruses that we see in tests from Asia and Europe is different than the types we're seeing in America," said Gail Rosen, PhD, a professor in Drexel's College of Engineering, who led the development of the tool. "Identifying the variations allows us to see how the virus has changed as it has traveled from population to population. It can also show us the areas where social distancing has been successful at isolating COVID-19."
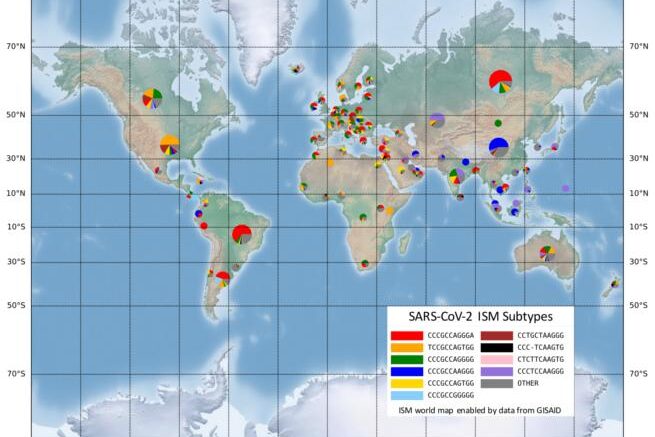
The ISM tool is particularly useful because it does not require analysis of the full genetic sequence of the virus to identify its mutations. In the case of SARS-CoV-2, this means reducing the 30,000-base-long genetic code of the virus to a subtype label 20 bases long.
The ISM tool also identified certain positions in the virus's genetic sequence that changed together as the virus spread. The researchers found that from early April to the end of the summer, three positions in the SARS-CoV-2 sequence mutated at the same time. Those positions are in different parts of the genetic sequence. One part is thought to be associated with cellular signaling and replication. Another portion is associated with formation of the protein spike - the part of the virus that enables its entry into healthy cells - changed in tandem with a third portion of code, which doesn't translate to protein.
While more investigation is needed on how these simultaneous mutations impact the transmission and severity of the virus, sites that change together can be used to consolidate the subtype label into 11 bases, which could make downstream analysis more efficient, according to the researchers.
"It's the equivalent of scanning a barcode instead of typing in the full product code number," Rosen said. "And right now, we're all trying to get through the grocery store a bit faster. For scientists this means being able to move to higher-level analysis much faster. For example, it can be a faster process in studying which virus versions could be affecting health outcomes. Or, public health officials can track whether new cases are the result of local transmission or coming from other regions of the United States or parts of the world."
While these genetic differences might not be enough to delineate a new strain of virus, Rosen's group suggests understanding these genetically significant "subtypes," where they're being found and how prevalent they are in these areas is data granular enough to be useful.
"This allows us to see the very specific fingerprint of COVID-19 from each region around the world, and to look closely at smaller regions to see how it is different," Rosen said. "Our preliminary analysis, using publicly available data from across the world, is showing that the combination of subtypes of virus found in New York is most similar to those found in Austria, France and Central Europe, but not Italy. And the subtype from Asia, that was detected here early in the pandemic has not spread very much, instead we are seeing a new subtype that only exists in America as the one most prevalent in Washington state and on the west coast."
In addition to helping scientists understand how the virus is changing and spreading, this method can also reveal the portion of its genetic code that appears to remain resistant to mutations - a discovery that could be exploited by treatments to combat the virus.
"We're seeing that the spike protein and the part of the virus responsible for packaging its genetic material have developed a few major mutations, but otherwise they are changing at a slower rate," said Bahrad Sokhansanj, PhD, a visiting scholar at Drexel. "Importantly, both are key targets for understanding the body's immune response, identifying antiviral therapeutics, and designing vaccines.
Rosen's Ecological and Evolutionary Signal-Processing and Informatics Laboratory will continue to analyze COVID-19 data as it is collected and to support public health researchers using the ISM process.
Source: Drexel University
515644 632684Satisfying posting. It would appear that a great deal of the stages are depending upon the originality aspect. Its a funny thing about life in the event you refuse to accept anything but the best, you very often get it. by W. Somerset Maugham.. 199237