Pathogens such as SARS-CoV-2 and pneumococcus can cause severe pneumonia. If the airways then fill with fluid, the patient risks developing acute respiratory distress syndrome. Researchers at Charité – Universitätsmedizin Berlin have now discovered the molecular mechanisms that trigger fluid accumulation in the lungs. This also led them to discover a potential new therapy: A cystic fibrosis drug proved effective in their laboratory experiments, raising hope that this could be used to treat pneumonia regardless of the pathogen that caused it. The study has been published in the journal Science Translational Medicine.
Pneumonia is the most common cause of fluid buildup in the lungs. This condition, known as pulmonary edema, results in parts of the airspaces filling with fluid instead of air, which prevents them from doing their job of exchanging gases. Patients struggle to breath and their body can’t get enough oxygen. The diagnosis is acute respiratory distress syndrome, or ARDS. “Despite cutting-edge medical procedures, roughly 40 percent of patients with ARDS die in intensive care. The problem is that antibiotics, antivirals, and immune modulating therapies rarely work well enough,” says study leader Dr. Wolfgang Kuebler, director of the Institute of Physiology at Charité. “That’s why we took a very different approach in our study. Instead of focusing on the pathogen, we focused on strengthening the barrier function of the blood vessels in the lungs.” This makes sense, as they are the source of the fluid in pulmonary edema. The lung vessels become permeable, allowing fluid from the blood to flow into the surrounding tissue – and thereby flood the airspaces.
But what actually causes this? What are the underlying molecular mechanisms? A Charité research team led by Kuebler set out to answer these questions. They performed experiments using cells, lung tissue, and isolated lungs. The study centered on the CFTR chloride channel, which scientists know is mainly found in the mucosal cells of our airways. There, it plays a major role in keeping our mucus thin so it can drain away easily. The researchers have now shown for the first time that cells in the blood vessels of the lungs also have CFTR and that its presence is drastically reduced in pneumonia.
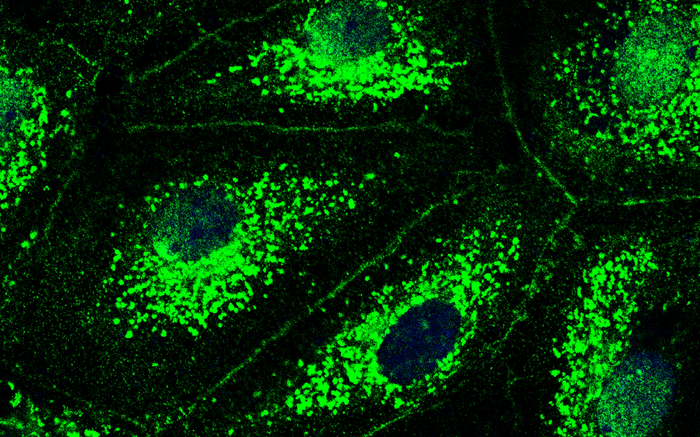
To find out what role CFTR plays in the pulmonary vessels and what is happening at the molecular level when the chloride channel is lost, the researchers blocked the channel with an inhibitor and dictated the number of chloride ions in the cells. They then used a special imaging technique known as immunofluorescence imaging: “We saw that inhibiting CFTR triggered a molecular cascade that ultimately causes the lung’s blood vessels to begin leaking,” says Dr. Lasti Erfinanda, who also works at the Institute of Physiology and is the study’s lead author. “So CFTR actually does play a very key role in the development of pulmonary edema.”
The study findings indicate that the loss of CFTR causes chloride to accumulate in the cells because it stops being transported out of them. The excess chloride triggers signaling that ends with an uncontrolled flow of calcium into the cells via a calcium channel. “The increased calcium concentration then causes the vascular cells to contract – much like the effect that calcium has on muscle cells,” explains Kuebler. “This results in gaps between the cells – which allows fluid to spill out of the blood vessels. Chloride channels are therefore crucial in maintaining the barrier function of the pulmonary vessels.”
The research team then addressed another question: How could they attenuate or prevent the pneumonia-induced loss of chloride channels in the pulmonary vessels? To answer this, the researchers used a therapeutic agent that is classed as a CFTR modulator and currently used to treat cystic fibrosis. In cystic fibrosis patients, a genetic mutation prevents the CFTR chloride channel from working properly in the mucosal cells of the airways, resulting in very viscous mucus. “Ivacaftor is a drug that increases the chances of the chloride channel opening, which helps the mucus to flow through the airways,” says Erfinanda. “We wanted to see if it would also have a positive effect on the cells in the blood vessels of the lungs.”
Ivacaftor did make the chloride channels more stable: it led to less degradation in the channels than that typically caused by the lung’s inflammatory processes. Experiments on animal models showed the same effect: treatment with ivacaftor increased the probability of surviving severe pneumonia, reduced lung injury, and resulted in much milder symptoms and a much better general condition than without the drug. “We really weren’t expecting it to work so well,” says Kuebler. “We hope our findings will pave the way for clinical trials to test the efficacy of CFTR modulators in pneumonia patients. If this promising, pathogen-independent therapy finds its way into clinical practice, it could benefit a huge number of patients and prevent pneumonia from becoming life-threatening – even in the case of unknown pathogens.”
Kuebler and his team are now planning research projects aimed at developing other potential therapies based on the CFTR signaling pathway. They are also going to research which patients have an elevated risk of developing ARDS, so they can provide these patients with preventive, personalized treatment.
Reference: Erfinanda L et al. Loss of endothelial CFTR drives barrier failure and edema formation in lung infection and can be targeted by CFTR potentiation. Science Translational Medicine 2022 Dec 07. doi: 10.1126/scitranslmed.abg8577ggf.