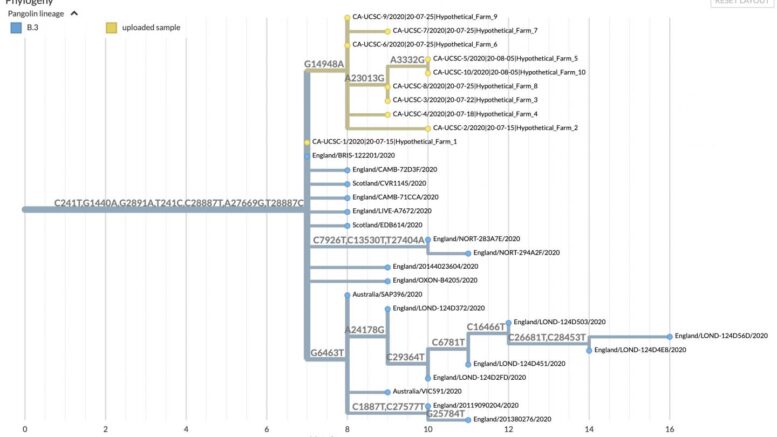
In this example of UShER results, displayed using Nextstrain, sequences representing a hypothetical outbreak are yellow, previously sampled sequences are blue, and branches are labeled by nucleotide mutations. Courtesy of UC Santa Cruz Genomics Institute
The COVID-19 pandemic has spurred genomic surveillance of viruses on an unprecedented scale, as scientists around the world use genome sequencing to track the spread of new variants of the SARS-CoV-2 virus. The rapid accumulation of viral genome sequences presents new opportunities for tracing global and local transmission dynamics, but analyzing so much genomic data is challenging.
"There are now more than a million genome sequences for SARS-CoV-2. No one had anticipated that number when we started sequencing this virus," said Russ Corbett-Detig, assistant professor of biomolecular engineering at UC Santa Cruz.
The sheer number of coronavirus genome sequences and their rapid accumulation makes it hard to place new sequences on a "family tree" showing how they are all related. But Corbett-Detig's group at the UC Santa Cruz Genomics Institute has developed a new method that does this with unprecedented speed. Called Ultrafast Sample Placement on Existing Trees (UShER), this powerful tool is described in a paper published May 10 in Nature Genetics.
UShER identifies the relationships between a user's newly sequenced viral genomes and all known SARS-CoV-2 virus genomes by adding them to an existing phylogenetic tree, a branching diagram like a family tree that shows how the virus has evolved in different lineages as it accumulates mutations.
"We are able to maintain a comprehensive phylogenetic tree of more than 1.2 million coronavirus sequences and update it with new sequences in real time. No other tool can handle trees of this size with a comparable efficiency," said first author Yatish Turakhia, a postdoctoral scholar at the Genomics Institute. "This helps us keep track of all variants in circulation, including new variants that are emerging."
This kind of sequence analysis can be used to discover new strains of the virus as they emerge and track their evolution and transmission dynamics. It can also be used to identify links between individual cases of coronavirus infection and to trace chains of transmission, an approach known as genomic contact tracing.
"The challenge is to get results soon enough to make meaningful predictions that public health agencies can use to try to control an outbreak," said Corbett-Detig, a corresponding author of the paper. "Our method is orders of magnitude faster than anything else out there, placing new samples in tenths of a second."
UShER and related data visualization tools are available to the research community through the UCSC SARS-CoV-2 Genome Browser, which also provides access to a wide range of data and results from ongoing scientific research on the virus, including new variants that are especially concerning.
"Our browser is the most comprehensive information resource for mutations appearing in the virus and what they mean for our battle against it," said coauthor David Haussler, professor of biomolecular engineering and director of the Genomics Institute. "Thanks to Russ's team, it includes the world's most comprehensive phylogenetic tree of the different lineages of the virus, and that tree continues to grow every week, as fast as new data appear."
Like all viruses, SARS-CoV-2 acquires mutations as it replicates and spreads. Most of these random variations in the genome sequence have no effect on the behavior of the virus, but researchers can still use them to identify different variants or strains of the virus, see how they are related, and determine if two samples are part of the same transmission chain.
Scientists have identified several important mutations that appear to make the virus more infectious. Variants of SARS-CoV-2 with these mutations are spreading more rapidly than other variants. Coauthor Angie Hinrichs, a UCSC Genome Browser engineer, used UShER to determine that one of these variants, known as B.1.1.7, entered the United States through several independent introductions. It is now the dominant strain in the United States.
Turakhia said he has begun using UShER to study a new variant that has emerged in India and appears to be spreading rapidly there. Known as B.1.617, this lineage of the virus has two mutations of potential concern to scientists. "We don't know yet how concerning it is, but it is important to track it," he said.
Viral genomics can reveal transmission chains not found through conventional contact tracing, Corbett-Detig said. This approach can help identify superspreader events, where one person transmitted the virus to many others, and it can also show that two cases from the same location are actually unrelated infections, not part of the same transmission chain, because the viral sequences differ too much.
"It's an approach that is likely to be valuable moving forward, so we're building the tools to enable people to do this in real time," he said. "If you want to know who transmitted the virus to whom, or where in the world a new sample may have come from, you need to take the samples from your community and project them onto the known phylogenetic tree of all the other SARS-CoV-2 genome sequences, and conventional phylogenetic methods just can't do this in a reasonable amount of time."
That's because conventional methods have to recalculate the entire tree every time new sequences are added, which is much too time-consuming when there are hundreds of thousands of sequences. UShER places samples onto an existing global phylogeny almost instantly, and it provides a local subtree of the added samples and their nearest neighbors so that their relationships can be visualized and examined in detail.
The researchers showed that UShER finds the right placement in 97% of cases. In the other 3%, incorrect placements are very close to the true site and still useful for contact tracing. UShER can also be used for quality control to quickly identify and remove low-quality sequences that may contain sequencing errors. The UShER results can be visualized and explored on the Nextstrain platform for interactive visualization of phylogenetic trees and maps of how the virus is spreading.
In addition to Turakhia, Corbett-Detig, Hinrichs, and Haussler, the coauthors of the paper include Bryan Thornlow and Landen Gozashti at UC Santa Cruz, Nicola De Maio at the European Bioinformatics Institute, and Robert Lanfear at Australian National University, Canberra. This work was funded by the Alfred P. Sloan Foundation and the National Institutes of Health.
Source: UC Santa Cruz
Be the first to comment on "New Tools Enable Rapid Analysis of Coronavirus Sequences and Tracking of Variants"