In a paper in the journal Nature, Los Alamos National Laboratory scientists Bette Korber, Hyejin Yoon, Will Fischer and James Theiler, among nearly 130 authors from institutions around the world, describe their groundbreaking collaborative work, “Defining the risk of SARS-CoV-2 variants on immune protection.”
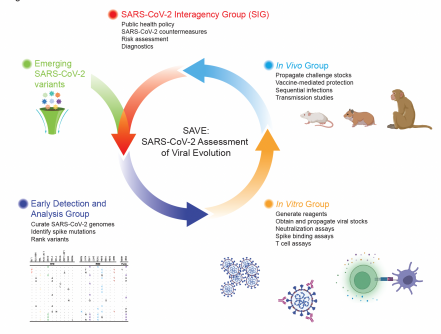
Korber, Fischer, Yoon and Theiler are members of a rarified team that the National Institute of Allergy and Infectious Diseases assembled in January 2021, drawing on experts from around the world who specialize in relevant research fields such as viruses, the immune system, vaccines, epidemiology, structural biology, bioinformatics, virus genetics, and evolution. The team is called SAVE, for SARS-CoV-2 Assessment of Viral Evolution.
As noted in the Nature paper, the authors state, “This effort was designed to provide a real-time risk assessment of SARS-CoV-2 variants potentially impacting transmission, virulence, and resistance to convalescent and vaccine-induced immunity. The SAVE program serves as a critical data-generating component of the United States Government SARS-CoV-2 Interagency Group to assess implications of SARS-CoV-2 variants on diagnostics, vaccines and therapeutics and for communicating public health risk.”
SAVE focuses on mutations in SARS-CoV-2 and emerging virus variants. But its members say the global collaborative concept “is a broad model for rapidly responding to evolving pathogens with pandemic potential.”
“Over the past two decades, we have witnessed the emergence/re-emergence of several RNA viruses, including West Nile virus, H1N1 influenza virus, chikungunya virus, Zika virus, SARS-CoV-1, MERS-CoV and Ebola virus, that have threatened global public health,” the paper’s summary states. “Developing collaborative programs between academic, industry and commercial partners is essential to respond to rapidly evolving viruses,” said Marciela DeGrace of NIAID, the paper’s lead author.
SAVE members represent 58 different research sites located in the United States and around the world. Members participate within three sub-groups:
- Early Detection and Analysis
- In Vitro – what they can learn using flasks, beakers and tubes
- In Vivo – what they can learn in animal models that mimic human disease
Korber’s team was part of the Early Detection and Analysis team, where such high-impact work as the initial identification of mutations in the virus made waves in the scientific community before its capacity for mutation had been clearly understood and accepted.
The Nature paper notes, “The process is collaborative and iterative, with seven teams using independent models and methodologies to prioritize mutations and lineages as well as rank importance for downstream testing. While the focus is on human infections, the Early Detection group also monitors variants circulating in animal populations, such as mink and deer, since they represent a potential reservoir source.”
On a weekly basis, the SAVE Early Detection and Analysis team reviews downloads of SARS-CoV-2 genomes from the international initiative for sequence sharing, GISAID. They search for variant and co-variant signatures in the genomes, then divide the work into two approaches:
- one based on convergent evolution as the main signal for selection and functional impact of mutations (done by Cambridge and Walter Reed Army Institute of Research teams)
- the other anchored on prevalence and growth patterns of mutations and defined lineages (the role of Los Alamos, Icahn School of Medicine at Mount Sinai, J. Craig Venter Institute/Bacterial Viral Bioinformatic Resource Center, UC-Riverside and Broad Institute teams)
At Los Alamos, the Korber team identifies emergent mutational patterns within the SARS-CoV-2 spike protein to track newly emerging and expanding variants and determine transitions in global and regional sampling frequencies over time, which is the specialty area in which Los Alamos has made a huge impact.
They pay particular attention to mutations in parts of the spike protein known to be highly targeted by antibodies, or that might impact infectivity. They also systematically define the most commonly circulating form of each emerging variant of interest or concern against the backdrop of the continuously evolving virus.
“Identifying the emerging variants, and obtaining accurate sequences for those variants, required continued wrangling of burgeoning data,” said Theiler. “There are now close to 10 million SARS-CoV-2 sequences in GISAID. These sequences, however, are non-uniformly sampled, are often partial and some contain errors, and of course it is the newest variants that give the sequencers the most trouble.”
“The tools we developed, along with our colleagues on the LANL COVID-19 Viral Genome Analysis Pipeline (cov.lanl.gov), provided the infrastructure that enabled us to follow this pandemic though its various waves,” he added.
Korber noted that “by working with the SAVE Early Detection team, we were able to be part of a synergistic collaborative effort, where our results in terms of early detection could be cross-checked with those of others.”
She added, “The real beauty of being part of the larger SAVE project was the knowledge that our analysis pipeline could provide foundational support for the many experimental teams in SAVE, and that we could help the scientific community get the best version of newly emergent variants into their laboratories as quickly and accurately as possible. In this way the science needed to understand the immunological and virological characteristics of new variants was rapidly obtained, in time to help inform public health decisions.”
Reference: M DeGrace, et al. Defining the risk of SARS-CoV-2 variants on immune protection. Nature https://doi.org/10.1038/s41586-022-04690-5
LA-UR-22-23006
Source: Los Alamos National Laboratory