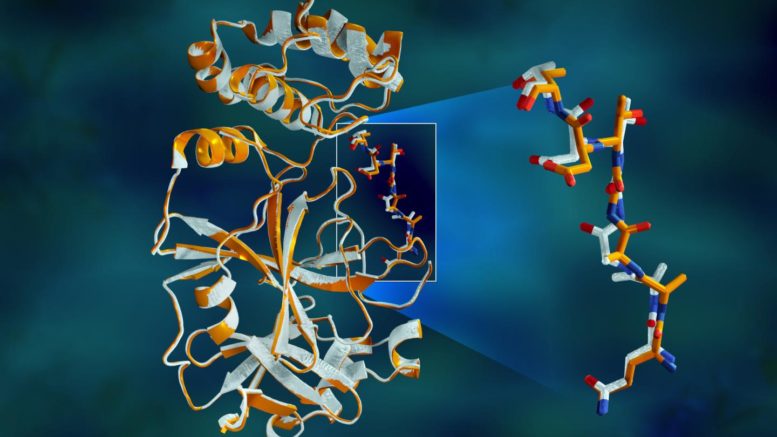
Overlapping X-ray data of the SARS-CoV-2 main protease shows structural differences between the protein at room temperature (orange) and the cryogenically frozen structure (white). Courtesy of Jill Hemman/ORNL, U.S. Dept. of Energy
A team of researchers at the Department of Energy's Oak Ridge and Argonne national laboratories has performed the first room-temperature X-ray measurements on the SARS-CoV-2 main protease -- the enzyme that enables the virus to reproduce.
The X-ray measurements mark an important first step in the researchers' ultimate goal of building a comprehensive 3D model of the enzymatic protein. The model will be used to advance supercomputing simulations aimed at finding drug inhibitors to block the virus's replication mechanism and help end the COVID-19 pandemic. Their research results are publicly available and have been published in the journal Nature Communications.
SARS-CoV-2 is the virus that causes the disease COVID-19. The virus reproduces by expressing long chains of proteins that must be cut into smaller lengths by the protease enzyme.
"The protease is indispensable for the virus life-cycle. The protein is shaped like a valentine's heart, but it really is the heart of the virus that allows it to replicate and spread. If you inhibit the protease and stop the heart, the virus cannot produce the proteins that are essential for its replication. That's why the protease is considered such an important drug target," said ORNL's Andrey Kovalevsky, corresponding author. While the structure is known from cryogenically preserved crystals, "This is the first time the structure of this enzyme has been measured at room temperature, which is significant because it's near the physiological temperature where the cells operate."
Building a complete model of the protein structure requires identifying each element within the structure and how they are arranged. X-rays are ideal for detecting heavy elements such as carbon, nitrogen and oxygen atoms. Because of the intensity of the X-ray beams at most large-scale synchrotron facilities, biological samples typically must be cryogenically frozen to around 100 K, or approximately minus 280 degrees Fahrenheit, to withstand the radiation long enough for data to be collected.
To extend the lifetime of the crystallized protein samples and measure them at room temperature, ORNL researchers grew crystals larger than required for synchrotron cryo-studies and used an in-house X-ray machine that features a less intense beam.
"Growing protein crystals and collecting data is a tedious and time-consuming process. In the time it typically takes to prepare and ship the sample to a synchrotron, we were able to grow the crystals, take the measurements and begin analyzing the data," said ORNL's Daniel Kneller, the study's first author. "And, when there's a pandemic with many scientists mobilizing to study this problem, there's not a day to spare."
The protease enzyme consists of chains of amino acids with a repeating pattern of nitrogen-carbon-carbon atoms that form the backbone of the protein. Side groups of the amino acid building blocks, or "residues", extend from each of the central backbone carbon atoms. The enzyme is folded into a specific 3D shape, creating special pockets where a drug molecule would attach.
The study revealed significant structural disparities between the orientations of the backbone and some of the residues in the room-temperature and cryogenic samples. The research suggests that freezing the crystals may introduce structural artifacts that could result in a less accurate understanding of the protease structure.
The team's results are being shared with researchers, led by ORNL-University of Tennessee Governor's Chair Jeremy Smith, who are conducting drug docking simulations using Summit at ORNL -- the nation's fastest supercomputer.
"What researchers are doing on Summit is taking known drug compounds and trying to computationally bind them to the main protease for drug repurposing, as well as looking for new leads into other potential drug candidates," said ORNL corresponding author Leighton Coates. "Our room temperature data is being used to build a more accurate model for those simulations and improve drug design activities."
The researchers' next step in completing the 3D model of the SARS-CoV-2 main protease is to use neutron scattering at ORNL's High Flux Isotope Reactor and the Spallation Neutron Source. Neutrons are essential in locating the hydrogen atoms, which play a critical role in many of the catalytic functions and drug design efforts.
The protease plasmid DNA used to make the enzyme was provided by Argonne's Structural Biology Center at the Advanced Photon Source. Crystallization of the proteins used in the X-ray scattering experiments was performed at ORNL's Center for Structural and Molecular Biology.
In addition to Kovalevsky, Kneller, and Coates, the paper's authors are ORNL's Gwyndalyn Phillips, Hugh M. O'Neill and Paul Langan; and Argonne's Robert Jedrzejczak, Lucy Stols and Andrzej Joachimiak.
The work was funded by ORNL's Laboratory Directed Research and Development program, the National Institute of Allergy and Infectious Diseases of the National Institutes of Health, and the National Science Foundation, with facilities support from DOE's Office of Science.
Be the first to comment on "X-rays Size Up Protein Structure at the ‘Heart’ of SARS-CoV-2"