Developing new drugs that remain potent against emerging variants of COVID-19 is essential to stopping the spread of the highly contagious disease and protecting human health.
After more than two years studying SARS-CoV-2—the virus that causes COVID-19—researchers at the Department of Energy’s (DOE) Oak Ridge National Laboratory (ORNL) are now designing and testing small-molecule antivirals that block the virus’s ability to reproduce. Along with their collaborators at the Institut Laue-Langevin (ILL), France, the researchers showed that their antiviral molecules are similarly as effective as some of the leading drugs on the market today.
The antiviral molecules, called hybrid inhibitors, are made from repurposed drugs used to treat hepatitis C and SARS-CoV—the coronavirus that broke out in 2002.
The team’s encouraging research results, published in the journal Nature Communications, indicate the novel antiviral molecules are worth further development for potential use as new drugs to treat various forms of the virus.
“From the beginning, our research has revolved around the main protease, which is an enzyme inside SARS-CoV-2 that enables the virus to reproduce. If you shut down the protease, you shut down the virus,” said Daniel Kneller, the paper’s lead author.
The SARS-CoV-2 main protease is shaped like a Valentine’s heart. On its surface are pockets that can bind long chains of amino acids expressed by the virus. The protease is responsible for cutting, or dividing, those chains, which is how the virus reproduces.
To better understand how the protease works, the researchers used a combination of X-rays and neutron experiments at ORNL’s Spallation Neutron Source (SNS) and High Flux Isotope Reactor (HFIR) to create a comprehensive 3-dimensional map of every atom in the protease structure. Next, they charted the vast network of hydrogen bonds that hold the protease together.
They also plotted the location of each pocket where the cutting process occurs and discovered the sites where the amino acid chains get cut are electrically charged. Knowing the positive, negative, and neutral electrical charges of the amino acid sites is key to designing antiviral molecules that bind tightly to the protease structure. The more tightly the molecules bind, the more effective they are at stopping the protease.
With the experimental data in hand, the researchers pivoted to investigating antiviral hepatitis C drugs that could potentially be redesigned, or repurposed, to block the SARS-CoV-2 protease. The team studied three hepatitis C drugs approved by the Federal Drug Administration—boceprevir, narlaprevir, and telaprevir. Neutron scattering experiments revealed that the SARS-CoV-2 protease has the unique ability to shape-shift and alter its electrical charge states to conform to whatever drug molecule is being introduced. The unforeseen discovery had not been predicted by computer simulations and provided another key piece of insight into designing new drug molecules that bind specifically to the target.
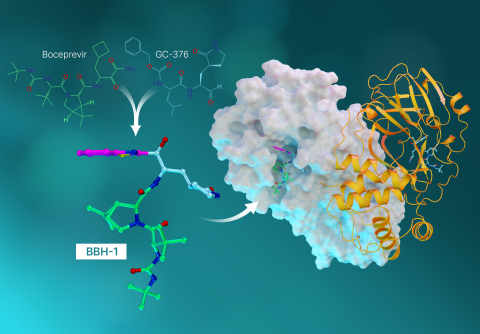
“This study is the culmination of everything we’ve learned up to this point. In it, we took the best parts of the hepatitis C drugs and created three new molecules and tested them on SARS-Cov-2’s protease,” said Kneller.
Each drug molecule has what’s called a warhead that links directly to the amino acid site where the natural chemical reaction would occur. Ideally, when the warhead binds to the amino acid site, the resulting combination of the enzyme and the inhibitor should closely represent the natural one.
“What we found in our previous studies is that when the telaprevir binds, the warhead has a neutral charge and actually faces away from the special amino acid site where interactions would be strong,” said ORNL senior scientist Andrey Kovalevsky. “So this time, we designed hybrid inhibitors to provide the exact linkage we needed to establish tight interactions between the inhibitors and the protease.”
This time, neutron scattering experiments were conducted using the LADI-DALI beamline at the ILL nuclear reactor to expedite the measurements of the molecular binding interactions between the three hybrid inhibitors and the SARS-CoV-2 protease. Because neutrons are nondestructive and highly sensitive to light elements such as hydrogen, they provide a powerful tool for studying complex biological processes.
“Neutrons enabled us to see for the first time these very strong binding interactions the hybrid molecules form with the protease. These specific features of their design could be incorporated in the development of new drugs that would potentially be more effective against viral replication,” said ILL instrument scientist Matthew Blakeley. “No other experimental technique can provide this level of detail, which is precisely what is needed to truly understand how these molecules work.”
Additional experiments were performed by researchers at the National Institutes of Health using in vitro enzyme kinetics to study the inhibitors in test tube solutions. These tests also confirmed strong binding interactions, which were closely comparable to some of the leading COVID-19 drugs now on the market.
Samples of the inhibitor molecules and SARS-CoV-2 protease used in the experiments were synthesized and developed at ORNL’s Center for Nanophase Materials Sciences (CNMS) and Center for Structural Molecular Biology.
“We’ve shown tremendous capabilities in our ability to provide fundamental knowledge about an emerging disease and apply that to expediting the timeline to realizing effective treatments,” said Kovalevsky. “This is a big step forward not only in fighting COVID-19, but hopefully our research will translate to similar challenges we may face in the future.”
In addition to Kneller, Blakeley, and Kovalevsky, the paper’s co-authors include Hui Li, Gwyndalyn Phillips, Kevin L. Weiss, Qiu Zhang, Mark A. Arnould, Colleen B. Jonsson, Surekha Surendranathan, Jyothi Parvathareddy, Leighton Coates, John M. Louis, and Peter V. Bonnesen.
Source: Oak Ridge National Laboratory